


© Alexander Raths/Fotolia
Hauptinhalt
Arzneimittel, Pharmazie
Arzneimittel
Die Bezirksregierung Münster überwacht den Verkehr mit Arzneimitteln und Wirkstoffen. Interessant ist, welche Produkte dabei unter den Begriff „Arzneimittel“ fallen:
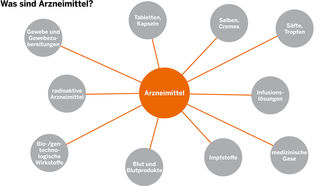
Das sind nicht nur die Präparate, die in der Apotheke erhältlich sind oder vom Arzt verabreicht werden, wie Tabletten, Kapseln, Salben, Cremes, Husten-Säfte, Tropfen, Impfstoffe und Infusionslösungen. Sondern auch Produkte, die auf den ersten Blick nicht als Arzneimittel erkannt werden.
Das Arzneimittelgesetz gilt zum Beispiel auch für medizinische Gase, bio- und gentechnologisch hergestellte Wirkstoffe, Blut und Blutprodukte, radioaktive Arzneimittel, Gewebe und Gewebezubereitungen wie Knochen, Gefäße und Augenhornhäute.
Zu den Überwachungsaufgaben der Bezirksregierung gehören die:
- Klinische Prüfung von Arzneimitteln
- Herstellung von Arzneimitteln und Wirkstoffen
- Inverkehrbringen von Arzneimitteln
- Handel mit Arzneimitteln und Wirkstoffen
- Import von Arzneimitteln und Wirkstoffen
- Export von Arzneimitteln
- Sicherheit von Arzneimitteln
Weitere Tätigkeiten, die im Zusammenhang mit Arzneimitteln stehen und bei der Bezirksregierung anzuzeigen sind, werden in § 67 des Arzneimittelgesetzes (AMG) aufgeführt.
Regelmäßige Inspektionen
Die entsprechenden Firmen und Einrichtungen werden in regelmäßigen Abständen und aus besonderen Anlässen, beispielsweise bei Änderung der Erlaubnis oder Bedenken gegen die Arzneimittelsicherheit, inspiziert.
Mit den Aufgaben und der Verantwortung, die die Bezirksregierung Münster im Rahmen der Routineüberwachung im Arzneimittelbereich und bei der Gefahrenabwehr übernimmt, trägt sie entscheidend zur Sicherheit der Patienten bei.
Für alle Belange, die Tierarzneimittel betreffen, ist in Nordrhein-Westfalen das Landesamt für Natur, Umwelt und Verbraucherschutz NRW (LANUV) zuständig.
Für die Zulassung von Arzneimitteln ist das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) zuständig, bei Blut und Blutprodukten ist es das Paul-Ehrlich-Institut (PEI) in Langen. Wird eine europaweite Zulassung eines Arzneimittels angestrebt, ist der Zulassungsantrag bei der „European Medicines Agency" (EMA) in London zu stellen.
Arzneimittelherstellung
Wer Arzneimittel im Sinne des § 2 Absatz 1 oder Absatz 2 Nr. 1 AMG, Testsera, Testantigene, Wirkstoffe, die menschlicher, tierischer oder mikrobieller Herkunft sind oder auf gentechnischem Wege hergestellt werden sowie andere zur Arzneimittelherstellung bestimmte Stoffe menschlicher Herkunft gewerbs- oder berufsmäßig herstellt, benötigt eine Herstellungserlaubnis gemäß § 13 Arzneimittelgesetz (AMG).
Zur Herstellung zählen das Gewinnen, Anfertigen, Zubereiten, Bearbeiten oder Verarbeiten, Umfüllen einschließlich Abfüllen, Abpacken, Kennzeichnen und die Freigabe von Arzneimitteln.
Die Erlaubnis für Humanarzneimittel ist vor Aufnahme der Tätigkeit bei der zuständigen Bezirksregierung zu beantragen. Für Tier-Arzneimittel ist das Landesamt für Natur, Umwelt und Verbraucherschutz NRW (LANUV) zuständig.
Eine Herstellungserlaubnis kann nur dann erteilt werden, wenn bestimmte personelle und sächliche Voraussetzungen erfüllt werden, die in den §§ 13 – 18 AMG näher bezeichnet sind. Im Zuge der Antragsbearbeitung werden diese auch im Rahmen einer Inspektion überprüft.
In einem Merkblatt (siehe Download) sind die Unterlagen, die bei einem Antrag auf Erteilung einer Herstellungserlaubnis vorzulegen sind, aufgelistet. Dort finden Sie auch Formulare, die bei der Antragstellung zu nutzen sind.
Die Erteilung der Herstellungserlaubnis ist mit einer Gebühr verbunden, die je nach Verwaltungsaufwand und wirtschaftlicher Bedeutung zwischen 100 und 25.500 Euro betragen kann.
Ärzte, Zahnärzte oder sonst zur Ausübung der Heilkunde am Menschen Befugte, unter deren unmittelbaren fachlichen Anleitung Arzneimittel zur persönlichen Anwendung hergestellt werden, haben dies bei der zuständigen Bezirksregierung anzuzeigen (Formular siehe Download. Das Formular ist mit einer Kopie der Approbationsurkunde oder der Anerkennung als Heilpraktiker/in zu ergänzen). Eine Erlaubnis ist für diese Tätigkeiten nicht erforderlich.
- Merkblatt für die Erteilung der Herstellungserlaubnis gemäß § 13 AMG (pdf, 244 KB) (Link öffnet sich in neuem Fenster)
- Anzeige für die Bestellung einer sachkundigen Person (pdf, 559 KB) (Link öffnet sich in neuem Fenster)
- Anzeige für die erlaubnisfreie Herstellung von Arzneimitteln durch Ärzte und zur Ausübung der Heilkunde bei Menschen durch befugte Personen (pdf, 121 KB) (Link öffnet sich in neuem Fenster)
Arzneimittelimport
Wer Arzneimittel im Sinne des § 2 Absatz 1 oder Absatz 2 Nr. 1 AMG, sowie andere zur Arzneimittelherstellung bestimmte Stoffe menschlicher Herkunft gewerbs- oder berufsmäßig aus Drittländern einführen will, benötigt eine Einfuhrerlaubnis gemäß § 72 Arzneimittelgesetz (AMG). Drittländer sind Länder, die nicht Mitgliedsstaaten der EU oder andere Vertragsstaaten des Abkommens über den Europäischen Wirtschaftsraum sind.
Privatpersonen ist die Einfuhr von Arzneimitteln aus Drittländern mit Ausnahme des Eigenbedarfes bei der Einreise nach Deutschland verboten.
Arzneimittel, die der Pflicht der Zulassung (§ 21 AMG) unterliegen, dürfen nur nach Deutschland eingeführt werden, wenn eine entsprechende Zulassung oder Genehmigung vorliegt. Die Erlaubnis ist vor Aufnahme der Tätigkeit bei der zuständigen Behörde zu beantragen.
Eine Einfuhrerlaubnis kann nur dann erteilt werden, wenn analog der Herstellungserlaubnis bestimmte personelle und sachliche Voraussetzungen erfüllt werden, die in den §§ 13 – 20a AMG näher bezeichnet sind (siehe Arzneimittelherstellung). Das Merkblatt fasst die Unterlagen zusammen, die auch bei einem Antrag auf Erteilung einer Einfuhrerlaubnis vorzulegen sind. Bei der Einfuhr von Arzneimitteln muss neben der Erlaubnis gemäß § 72 AMG auch ein Zertifikat gemäß § 72a AMG für den Hersteller der betreffenden Produkte vorliegen. Dazu ist in der Regel eine Inspektion der im Drittland liegenden Betriebsstätte durch die für den Einführer zuständigen Bezirksregierung erforderlich.
Für die zollamtliche Abfertigung ist eine Bescheinigung nach § 73 Absatz 6 AMG vorzulegen.
Die Erteilung der Einfuhrerlaubnis und der anderen erforderlichen Dokumente ist mit einer Gebühr verbunden:
- Einfuhrerlaubnis: je nach Verwaltungsaufwand und wirtschaftlicher Bedeutung 400 bis 25.500 Euro
- Gebührenrahmen für eine Auslandinspektion nach § 72a: derzeit 1.000 – 25.500 Euro.
- Ausstellung eines Zertifikates nach § 72a AMG: derzeit 130 – 390 Euro je Arzneimittel;
- Gebührenrahmen für die Bescheinigung nach § 73 Abs. 6 AMG: derzeit 130 – 500 Euro
Die Einfuhr von Fertigarzneimitteln, die nicht in Deutschland zugelassen sind, ist nur gestattet, wenn
- eine ärztliche Verschreibung für eine einzelne Person vorliegt,
- sie in geringer Menge von einer Apotheke bestellt und von dieser an den Patienten abgegeben werden,
- sie in dem Staat, aus dem sie eingeführt werden, rechtmäßig in Verkehr sind und
- in Deutschland hinsichtlich des Wirkstoffs identische und hinsichtlich der Wirkstärke vergleichbare Arzneimittel nicht zur Verfügung stehen.
Arzneimittelexport
Auf Antrag des pharmazeutischen Unternehmers, des Herstellers, des Ausführers oder der zuständigen Behörde des Bestimmungslandes stellt die Bezirksregierung Münster ein Zertifikat entsprechend dem Zertifikatsystem der Weltgesundheitsorganisation (WHO) aus.
Die Leitlinien zur Durchführung des Zertifikatsystems sind auf der Homepage der WHO abrufbar. Eine Mustervorlage für diese Zertifikate finden Sie unter auf der Homepage der ZLG. Die Ausstellung dieses Exportzertifikates ist gebührenpflichtig. (Gebührenrahmen derzeit 130 - 200 Euro für ein Arzneimittel)
Arzneimittelgroßhandel
Die Erlaubnis ist vor Aufnahme der Tätigkeit bei der zuständigen Behörde zu beantragen. Adressat für die Antragsstellung ist die zuständige Bezirksregierung.
Eine Großhandelserlaubnis kann nur dann erteilt werden, wenn bestimmte personelle und sächliche Voraussetzungen erfüllt werden.
Für die Anzeige der verantwortlichen Person gemäß § 52a AMG steht ein Formular zur Verfügung (siehe Download). Die Unterlagen, die bei einem Antrag auf Erteilung einer Großhandelserlaubnis vorzulegen sind, sind in einem Merkblatt (siehe Download) zusammengefasst.
Arzneimittelvermittlung
Für die Anzeige als Arzneimittelvermittler sind die in einem Merkblatt genannten Unterlagen einzureichen bei der
Bezirksregierung Münster
Dezernat 24
Domplatz 1 – 3
48143 Münster
Arzneimittelvertrieb / Pharmazeutischer Unternehmer
Für die Betriebe, die neben einer Erlaubnis gemäß § 13 oder § 52a AMG zusätzlich auch die Funktion eines pharmazeutischen Unternehmers (pU) wahrnehmen, sind neben der Anzeige weitere Unterlagen bei der zuständigen Bezirksregierung einzureichen. In einem Merkblatt sind die Unterlagen aufgelistet. Weitere Informationen zum Thema Pharmazeutischer Unternehmer finden Sie im Internet bei der Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten ZLG oder in der FAQ-Liste zu pharmazeutischen Unternehmern.
Wirk- und Hilfsstoffe
Betriebe und Einrichtungen, die Wirkstoffe oder andere zur Arzneimittelherstellung bestimmte Stoffe (Hilfsstoffe) herstellen, prüfen, lagern, verpacken, in den Verkehr bringen oder sonst mit ihnen Handel treiben müssen diese Tätigkeiten nach § 67 in Verbindung mit § 146 Absatz 10 des Arzneimittelgesetzes (AMG) anzeigen und unterliegen der Überwachung nach § 64 AMG.
Zuständige Behörde ist die Bezirksregierung, die entsprechende Firmen und Einrichtungen inspiziert und bei Erfüllung der rechtlichen Voraussetzungen arzneimittelrechtliche Zertifikate oder Erlaubnisse nach §§ 13 Absatz 1, 72 Absatz 1 AMG (erlaubnispflichtige Wirkstoffe menschlicher, tierischer oder mikrobieller Herkunft oder nach einem gentechnischen Verfahren gewonnene Wirkstoffe) erteilt.
Für die genannten Tätigkeiten gelten neben den Voraussetzungen des Arzneimittelgesetzes insbesondere folgende Vorgaben:
- Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV)
- EU-Leitfaden, insbesondere Teil II des EU-Leitfadens der Guten Herstellungspraxis für Wirkstoffe, einschließlich der Anhänge
- EU-Leitlinien zu den Grundsätzen der guten Vertriebspraxis für Wirkstoffe von Humanarzneimitteln
- EU-Leitlinien für die formalisierte Risikobewertung zur Ermittlung der angemessenen guten Herstellungspraxis für Arzneiträgerstoffe in Humanarzneimitteln
- Delegierte Verordnung (EU) Nr. 1252/2014 der Kommission zur Ergänzung der Richtlinie 2001/83/EG hinsichtlich der Grundsätze und Leitlinien der guten Herstellungspraxis für Wirkstoffe für Humanarzneimittel
Wirkstoffimport
Einfuhr von Wirkstoffen für Humanarzneimittel aus Drittländern, also aus Ländern, die nicht Mitgliedsstaaten der EU oder andere Vertragsstaaten des EWR sind. Generell gilt: Wer Wirkstoffe herstellt, prüft, lagert, verpackt, in den Verkehr bringt oder sonst mit ihnen Handel treiben will, muss dieses vor Aufnahme der Tätigkeit bei der zuständigen Behörde anzeigen. Neben den Regelungen des Arzneimittelgesetzes (AMG) sind insbesondere die Regelungen der Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) zu beachten.
- Wirkstoffe, die menschlicher, tierischer oder mikrobieller Herkunft sind oder auf gentechnischem Wege hergestellt werden
Wer diese Wirkstoffe gewerbs- oder berufsmäßig aus Drittländern einführen will, benötigt eine Einfuhrerlaubnis gemäß § 72 AMG. Die Erlaubnis ist vor Aufnahme des Importes von Wirkstoffen für Human-Arzneimittel bei der zuständigen Bezirksregierung zu beantragen. Für die Erteilung einer Erlaubnis für den Import von Wirkstoffen für Tier-Arzneimittel ist das Landesamt für Natur, Umwelt und Verbraucherschutz NRW (LANUV) zuständig.
Eine Einfuhrerlaubnis kann nur dann erteilt werden, wenn analog der Herstellungserlaubnis bestimmte personelle und sächliche Voraussetzungen erfüllt werden, die in den §§ 13 – 20a AMG näher bezeichnet sind, siehe Arzneimittelherstellung. Das dort eingestellte Merkblatt fasst die Unterlagen zusammen, die auch bei einem Antrag auf Erteilung einer Einfuhrerlaubnis vorzulegen sind.
Bei der Einfuhr von Wirkstoffen, die menschlicher, tierischer oder mikrobieller Herkunft oder auf gentechnischem Wege hergestellt sind, muss neben der Erlaubnis gemäß § 72 AMG auch ein Zertifikat gemäß § 72a AMG für den Hersteller der betreffenden Produkte vorliegen. Dazu ist in der Regel eine Inspektion der für den Einführer zuständigen Bezirksregierung der im Drittland liegenden Betriebsstätte erforderlich. Für Wirkstoffe menschlicher, tierischer oder mikrobieller Herkunft, die für ein nach einer im Homöopathischen Teil des Arzneibuches beschriebenen Verfahren zur Arzneimittelherstellung bestimmt, gelten diese Anforderungen gemäß § 72a Absatz 1a Nr. 3 AMG nicht.
Ein Zertifikat nach § 72a AMG und die Inspektion durch die Behörde im Drittland entfällt, wenn das Land, in dem der Herstellungsbetrieb liegt, in der von der Europäischen Kommission nach Art. 111b der RL 2001/83/EG veröffentlichten Liste aufgeführt ist (§ 72 a Absatz 1 Nr. 8 AMG).
Die Erteilung der Einfuhrerlaubnis ist mit einer Gebühr verbunden, die je nach Verwaltungsaufwand und wirtschaftlicher Bedeutung bis zu 25.500 Euro betragen kann. Der Gebührenrahmen für eine Auslandinspektion nach § 72a AMG liegt derzeit bei 1.000 – 25.500 Euro, für ein Zertifikat nach § 72a AMG Absatz 1 bei 130 – 390 Euro pro Arzneimittel.
- Wirkstoffe chemischen oder pflanzlichen Ursprungs
Seit Juli 2013 gilt: Wer Wirkstoffe chemischen oder pflanzlichen Ursprungs, die für die Herstellung von Human-Arzneimitteln vorgesehen sind, gewerbs- oder berufsmäßig aus Drittländern einführen will, benötigt ein Zertifikat der zuständigen Behörde des Herkunftslandes (Written Confirmation), in dem bestätigt wird, dass die Herstellung der Wirkstoffe nach anerkannten Grundregeln für die Herstellung und Sicherung der Qualität der EU oder nach anerkannten Standards (WHO, ICH Q 7a) erfolgt, die Herstellung regelmäßig überwacht wird, die Überwachung durch ausreichende Maßnahmen einschließlich wiederholter und unangekündigter Inspektionen erfolgt und bei wesentlichen Abweichungen von den Grundregeln die zuständige Behörde informiert wird. Dieses Zertifikat wird für jedes Arzneimittel einer Herstellungsstätte ausgestellt. Eine Kopie des betreffenden Zertifikates muss Bestandteil der Lieferpapieren der Sendung sein. Diese Anforderungen gelten nur für die Länder, die nicht in der von der Europäischen Kommission nach Artikel 111b der Richtlinie 2001/83/EG veröffentlichten Liste aufgeführt sind.
Blut und Blutprodukte
Die Herstellung und das Inverkehrbringen von Blut und Blutprodukten werden ebenfalls von der Bezirksregierung Münster überwacht. Dazu zählen unter anderem Fremdblut und daraus hergestellte Produkte, Eigenblut und periphere Blutstammzellen.
Für die Herstellung gelten die Voraussetzungen der §§ 13 - 18 Arzneimittelgesetz (siehe Arzneimittelherstellung). Darüber hinaus sind die besonderen Vorschriften des Transfusionsgesetzes (TFG) und die Richtlinien der Bundesärztekammer zur Gewinnung von Blut und Blutbestandteilen und zur Anwendung von Blutprodukten (Hämotherapie) zu beachten. Qualitätsmängel sind der Bezirksregierung Münster unverzüglich zu melden.
Für die Zulassung von Blutprodukten ist das Paul-Ehrlich-Institut (PEI) in Langen zuständig.
Weitere Informationen zum Thema „Blut- und Plasmaspende“ erhalten Sie beim Bundesministerium für Gesundheit und beim PEI.
- Gesetz zur Regelung des Transfusionswesens TFG (externer Link öffnet sich in neuem Fenster)
- Richtlinien zur Gewinnung von Blut und Blutbestandteilen und zur Anwendung von Blutprodukten (Hämotherapie) (externer Link öffnet sich in neuem Fenster)
- Anhang 14 zum EG-Leitfaden der Guten Herstellungspraxis (pdf, 49 KB) (Link öffnet sich in neuem Fenster)
Erlaubnispflicht für Gewebe und Gewebezubereitungen
Gewebe und Gewebezubereitungen wie zum Beispiel Herzklappen, Augenhornhäute, Knochen oder Blutgefäße unterliegen den Bestimmungen des Arzneimittelrechts. Dies gilt auch für menschliche Keimzellen, die im Rahmen der medizinisch unterstützten Befruchtung, unter anderem durch instrumentelle Samenübertragung oder intrazytoplasmatische Spermieninjektion (ICSI-Methode) Verwendung finden.
Herstellung von Gewebe und Gewebezubereitungen
Für folgende Tätigkeiten ist eine arzneimittelrechtliche Erlaubnis notwendig:
- Erlaubnis nach § 20b Arzneimittelgesetz (AMG)
- Gewinnen von Gewebe
- Laboruntersuchungen, die für die Gewinnung von Geweben erforderlich sind
- Erlaubnis nach § 20c Arzneimittelgesetz (AMG)
- Be- und Verarbeitung, Konservierung, Prüfung und Lagerung einschließlich Inverkehrbringen von Gewebe oder Gewebezubereitungen
Eine Erlaubnis ist auch dann erforderlich, wenn das Gewebe oder die Gewebezubereitung innerhalb der eigenen Einrichtung weitergegeben wird. Ausgenommen von der Erlaubnispflicht sind ausschließlich solche Gewebe, die innerhalb eines Behandlungsvorgangs einer Person entnommen werden, um auf diese ohne Änderung ihrer stofflichen Beschaffenheit rückübertragen zu werden (§ 4a Satz 1 Nr. 3 AMG).
Eine ärztliche Person, die die in § 20b oder § 20c AMG genannten Tätigkeiten mit Ausnahme des Inverkehrbringens ausübt, um das Gewebe oder die Gewebezubereitung persönlich bei ihren Patienten anzuwenden, hat dies bei der zuständigen Bezirksregierung anzuzeigen (Formular siehe Download-Bereich). Sie benötigt jedoch keine Erlaubnis nach § 20b oder § 20c AMG. Diese Freistellung von der Erlaubnispflicht gilt allerdings nicht für Arzneimittel, die zur klinischen Prüfung bestimmt sind (§ 20d AMG). Allerdings ist für Gewebe und Gewebezubereitungen an Stelle der Erlaubnis nach § 20c AMG eine Herstellungserlaubnis nach § 13 Absatz 1 AMG (siehe Herstellung von Arzneimitteln) dann erforderlich,
- wenn die Gewebe/Gewebezubereitungen mit industriellen Verfahren be- oder verarbeitet werden oder
- wenn wesentliche Be- oder Verarbeitungsverfahren in der EU nicht hinreichend bekannt sind.
Eine entsprechende Erlaubnis nach den §§ 20b oder 20c AMG kann nur dann erteilt werden, wenn bestimmte personelle und sachliche Voraussetzungen erfüllt werden. Diese sind in den § 20b und 20c AMG näher beschrieben. Informationen über die Unterlagen, die bei einem Antrag auf Erteilung einer Erlaubnis nach § 20b bzw. § 20c vorzulegen sind, sowie Formulare, die bei der Antragsstellung zu nutzen sind, finden Sie im Download.
Die Erteilung der jeweiligen Erlaubnis ist mit einer Gebühr verbunden, die je nach Verwaltungsaufwand und wirtschaftlicher Bedeutung zwischen 100 Euro und 25.500 Euro betragen kann.
Einfuhr von Geweben und Gewebezubereitungen
Für die Einfuhr von Gewebe und Gewebezubereitungen aus Drittstaaten, die nicht Vertragsstaaten des Abkommens über den europäischen Wirtschaftsraum sind, benötigt der Antragsteller darüber hinaus ein Zertifikat nach § 72b AMG. Dazu können, je nach Ermessen, eine Inspektion durch die Bezirksregierung in der außerhalb der EU ansässigen Einrichtung für die Gewinnung oder die Be- und Verarbeitung von Geweben oder Laboruntersuchungen notwendig werden. Die Erteilung der Einfuhrerlaubnis und des Zertifikats für die im Drittstaat befindliche Einrichtung ist ebenfalls mit einer entsprechenden Gebühr verbunden. Auch die Kosten für eine Inspektion der ausländischen Einrichtung gehen zu Kosten des Antragstellers.
- Gewebegesetz (externer Link öffnet sich in neuem Fenster)
- Merkblatt zur Erteilung einer Erlaubnis nach § 20b AMG (docx, 21 KB) (Link öffnet sich in neuem Fenster)
- Bestellung der angemessen ausgebildeten Person gemäß § 20b AMG sowie ärztlichen Person gemäß § 8 d TPG (docx, 17 KB) (Link öffnet sich in neuem Fenster)
- Merkblatt zur Erteilung einer Erlaubnis nach § 20c AMG (docx, 22 KB) (Link öffnet sich in neuem Fenster)
- Bestellung der verantwortlichen Person nach § 20c AMG (docx, 19 KB) (Link öffnet sich in neuem Fenster)
- Anzeige erlaubnisfreier Tätigkeiten § 13 Absatz 2b und § 20d AMG (pdf, 121 KB) (Link öffnet sich in neuem Fenster)
- Merkblatt zu externen Prüflaboren (pdf, 16 KB) (Link öffnet sich in neuem Fenster)
Klinische Prüfungen von Arzneimitteln
Die klinische Prüfung soll valide Daten zur Eignung und Sicherheit eines Arzneimittels liefern. Die menschliche Gesundheit ist ein hohes Gut. Deshalb hat der Schutz des Menschen während der klinischen Prüfung oberste Priorität. Gesetzliche Regelungen zur klinischen Prüfung von Arzneimitteln finden sich im sechsten Abschnitt des Arzneimittelgesetzes (§§ 40 – 42a AMG). Diese gesetzlichen Regelungen werden ergänzt und konkretisiert durch die aufgrund des AMG erlassene Verordnung zur "Guten Klinischen Praxis" (GCP-Verordnung). Letztere enthält weitere Einzelheiten für Sponsoren und Prüfer sowie die Verpflichtung zur Überwachung der Durchführung klinischer Studien durch die Behörden des Bundes und der Länder.
„Gute klinische Praxis" gemäß EU-Richtlinie 2001/20 Art. 1 Satz 2
Die gute klinische Praxis umfasst einen Katalog international anerkannter ethischer und wissenschaftlicher Qualitätsanforderungen, die bei der Planung, Durchführung und Aufzeichnung klinischer Prüfungen an Menschen sowie der Berichterstattung über diese Prüfungen eingehalten werden müssen. Die Einhaltung dieser Praxis gewährleistet, dass die Rechte, die Sicherheit und das Wohlergehen der Teilnehmer an klinischen Prüfungen geschützt werden und dass die Ergebnisse der klinischen Prüfungen glaubwürdig sind. Grundsätzlich sind alle Betriebe und Einrichtungen, die Arzneimittel am Menschen klinisch prüfen oder prüfen lassen, dazu verpflichtet, ihre Tätigkeit vor Beginn der zuständigen Überwachungsbehörde anzuzeigen. Neben Prüfeinrichtungen zählen hierzu insbesondere Sponsoren und durch diese beauftragte Auftragsforschungseinrichtungen (CROs). Zur Vereinfachung wurde hierzu ein bundeseinheitliches Anzeigenformblatt entwickelt.
Sponsor und CROs
In NRW sind die Anzeigen von Sponsoren und CROs (mit Sitz in NRW) an die jeweils zuständigen Bezirksregierungen zu richten. Die nordrheinwestfälischen Bezirksregierungen sehen in der Regel von der zusätzlichen Vorlage von Ethikvoten und Versicherungsnachweisen ab, sofern die Anzeige einer klinischen Prüfung auf dem bundeseinheitlichen Anzeigenformblatt erfolgt. Bei den Bezirksregierungen werden die Anzeigen gesammelt und bilden die Grundlage zur Vorbereitung von Inspektionen. Nach § 64 AMG unterliegen alle Parteien, die in die Studiendurchführung involviert sind, der Überwachung durch die zuständigen Behörden. Dazu gehören die Institutionen, die klinische Studien in Auftrag geben (Sponsoren) oder im Auftrag vornehmen (Auftragsforschungsinstitute oder CROs), Prüfer und auch in Studien involvierte Laboratorien. Zentrales Ziel der Inspektionen ist immer der Schutz der teilnehmenden Probandinnen und Probanden. Die Inspektionen erfolgen in der Regel mit Vorankündigung.
In einzelnen Fällen gibt es neben den Routineinspektionen auch anlassbezogene Inspektionen, die kurzfristig und unangekündigt erfolgen. Eine Inspektion gliedert sich in einen systembezogenen Teil (Teil I) und einen prüfungsbezogenen Teil (Teil II). Teil I befasst sich mit der strukturellen Organisation der Prüfeinrichtung. Dabei werden unter anderem Aufgaben und Verantwortlichkeiten von verschiedenen Abteilungen wie zum Beispiel Klinische Forschung, Arzneimittelsicherheit oder Qualitätssicherung hinterfragt. Teil II widmet sich im Wesentlichen der Überprüfung von Dokumenten konkreter klinischer Studien. In der Regel werden für eine Inspektion zwei Inspektionstage angesetzt. Eingebundene Laboratorien sollen zukünftig stärker in die Überwachung einbezogen werden. Voraussichtlicher Zeitumfang: ein Tag. Sämtliche GCP-Inspektionen unterliegen einem in der Verwaltungsgebührenordnung NRW festgesetzten Kostensatz. Die Gebühren orientieren sich am zeitlichen Aufwand für die jeweilige Überwachungsmaßnahme und sind von der jeweils inspizierten Einrichtung zu begleichen.
Prüfarzt
Eine Klinische Prüfung, zum Beispiel in einer Prüfeinrichtung oder bei einem Prüfarzt, ist nach den Bestimmungen der Verordnung über die Zuständigkeiten im Arzneimittelwesen und nach dem Medizinproduktegesetz NRW (Zuständigkeitsverordnung NRW) beim Zentralen Inspektorat für klinische Prüfstellen in Nordrhein-Westfalen anzuzeigen. Die Anzeigen sind an folgende Anschrift zu richten:
Landeshauptstadt Düsseldorf
Stadtverwaltung Amt 53
Zentrales Inspektorat für klinische Prüfstellen in NRW
40200 Düsseldorf
Arzneimittelsicherheit
Durch Qualitätsmängel von Arzneimitteln können Gefahren für die Gesundheit der Bevölkerung und die öffentliche Sicherheit und Ordnung entstehen. Bei unvorhergesehenem Auftreten dieser Mängel sind die notwendigen Maßnahmen einzuleiten und erforderlichenfalls auch länderübergreifend zu koordinieren.
Informationswege und Maßnahmen bei Qualitätsmängeln von Arzneimitteln sind im Erlass des Ministeriums für Arbeit, Gesundheit und Soziales NRW, Az. III C 4 - 0611.63.3 - vom 12.2.2008 inklusive 3 Anlagen geregelt.
Behörden und Institutionen
Deutsche und internationale Behörden
- Bundesministerium für Gesundheit (BMG) (externer Link öffnet sich in neuem Fenster)
- Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) (externer Link öffnet sich in neuem Fenster)
- Deutsches Institut für Medizinische Dokumentation und Information (DIMDI) (externer Link öffnet sich in neuem Fenster)
- Paul-Ehrlich-Institut (PEI) (externer Link öffnet sich in neuem Fenster)
- Food and Drug Administration (FDA) (externer Link öffnet sich in neuem Fenster)
- The European Agency for the Evaluation of Medicinal Products (EMEA) (externer Link öffnet sich in neuem Fenster)
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) (externer Link öffnet sich in neuem Fenster)
- European Directorate for the Quality of Medicines (EDQM) (externer Link öffnet sich in neuem Fenster)
- Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten (ZLG) (externer Link öffnet sich in neuem Fenster)
Fachverbände, Kammern, Informationszentralen
- Bundesverband der pharmazeutischen Industrie e.V. (BPI) (externer Link öffnet sich in neuem Fenster)
- Verband forschender Arzneimittelhersteller e.V. (VFA) (externer Link öffnet sich in neuem Fenster)
- Bundesfachverband der Arzneimittelhersteller e.V. (BAH-Bonn) (externer Link öffnet sich in neuem Fenster)
- Integritas - Verein für lautere Heilmittelwerbung e.V. (externer Link öffnet sich in neuem Fenster)
- Bundesverband des pharmazeutischen Großhandels e.V. (PHAGRO) (externer Link öffnet sich in neuem Fenster)
- Bundesverband medizinischer Auftragsinstitute e.V. (BVMA) (externer Link öffnet sich in neuem Fenster)
- Arzneimittelkommission der Deutschen Apotheker (AMK) (externer Link öffnet sich in neuem Fenster)
- Informationszentrale gegen Vergiftungen Universitätsklinikum Bonn (externer Link öffnet sich in neuem Fenster)
Rechtsvorschriften
- Arzneimittelgesetz (AMG) (externer Link öffnet sich in neuem Fenster)
- Allgemeine Verwaltungsvorschrift zur Durchführung des Arzneimittelgesetzes (AMGVwV) (pdf, 274 KB) (Link öffnet sich in neuem Fenster)
- Arzneimittelrecht Paul-Ehrlich-Institut (PEI) (externer Link öffnet sich in neuem Fenster)
- Arzneimittelhandelsverordnung (AM-HandelsV) (externer Link öffnet sich in neuem Fenster)
Verwandte Themen
Zusätzliche Informationen
Weitere Links
- ZLG - Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten (externer Link öffnet sich in neuem Fenster)
- Bundesgesundheitsministerium (externer Link öffnet sich in neuem Fenster)
- Paul-Ehrlich-Institut (externer Link öffnet sich in neuem Fenster)
- Robert-Koch-Institut (externer Link öffnet sich in neuem Fenster)
- Robert-Koch-Institut – FAQ Schutzimpfung gegen Masern (externer Link öffnet sich in neuem Fenster)